09.02.2017
Autorzy:
Praktyki:
Specjalizacje:
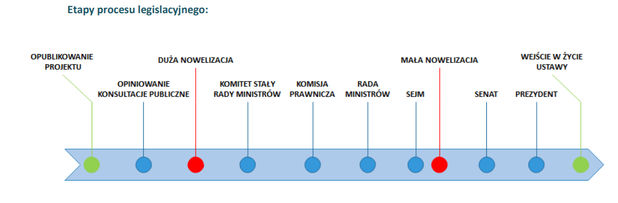
08.02.2017 do dalszej fazy uzgodnień przekazany został projekt tzw. Dużej nowelizacji ustawy o refundacji. Projekt jest bardzo podobny do propozycji analizowanej we wcześniejszych alertach.
W dniu 8 lutego br. do dalszej fazy uzgodnień przekazany został projekt ustawy o zmianie ustawy o refundacji leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych oraz niektórych innych ustaw – tzw. Duża nowelizacja ustawy o refundacji (DNUR). Przedmiotowy projekt jest w wielu miejscach bardzo podobny do propozycji analizowanej we wcześniejszych alertach. Zawiera jednak kilka istotnych zmian – część z nich ma na celu doprecyzowanie wcześniejszych propozycji, w przypadku innych Minister Zdrowia zdecydował o rezygnacji z poprzednio rozważanych przepisów.
Natomiast drugi z procedowanych obecnie projektów, tj. projekt ustawy o świadczeniach opieki zdrowotnej finansowanych ze środków publicznych oraz niektórych innych ustaw – tzw. Mała nowelizacja ustawy o refundacji (MNUR), został skierowany do Sejmu. Projekt wprowadza m.in. ratunkowy dostęp do technologii lekowych, jak również obejmuje zmiany ustawy o refundacji, w tym dotyczące między innymi okresu obowiązywania decyzji refundacyjnych.
W związku z powyższym wraz z PEX PharmaSequence Sp. z o.o. przedstawiamy Państwu zaktualizowany alert refundacyjny, w którym przeanalizowaliśmy najważniejsze zmiany prawne proponowane w obecnych projektach. Jednocześnie, przygotowując niniejszy alert dokonaliśmy analizy tego co zostało zmienione w w/w projektach w stosunku do tekstu, który był analizowany w trakcie konferencji refundacyjnej w dniu 12 września 2016 r. oraz we wcześniejszych alertach. W przypadku braku wskazania na takie zmiany tekst analizy pozostał niezmieniony.
Analizowane projekty należą do szeregu aktów prawnych, które zmieniły bądź mają w przyszłości zmienić system refundacji. Wśród przepisów, które weszły w życie należy przypomnieć regulacje związane z bezpłatnym dostępem do terapii dla pacjentów, którzy ukończyli 75. rok życia czy regulacje wprowadzające System Obsługi List Refundacyjnych (SOLR). Dodatkowo należy pamiętać o projekcie, który wprowadza szeroki zakres zmian w refundacji wyrobów medycznych dostępnych dotychczas na zlecenie bądź stosowanych w ramach realizacji świadczeń opieki zdrowotnej finansowanych ze środków publicznych.
Wniosek, który z tego płynie, wskazuje, że zamiast jednej kompleksowej zmiany prawnej będziemy mieli do czynienia z kilkoma aktami prawnymi, których wspólnym celem jest zmiana obecnego 2 systemu refundacji. W związku z tym odpowiedź na pytanie co czeka nas w 2017 r. wymaga kompleksowej analizy wszystkich wskazanych dotychczas projektów oraz obserwowania przyszłych prac toczących się przed Parlamentem.
Wracając do omówienia projektów będących powodem powstania niniejszego alertu należy wskazać, że zgodnie z uzasadnieniem przedstawionym przez autorów projektu DNUR, jego głównym celem jest wprowadzenie przeglądu oraz modyfikacji przepisów ustawy o refundacji. Zgodnie z tym samym uzasadnieniem, wynikiem przyjęcia proponowanych przepisów ma być wprowadzenie rozwiązań pozwalających na poprawę obowiązujących przepisów bądź usunięcie wątpliwości interpretacyjnych związanych z ich stosowaniem na przestrzeni ostatnich lat. Analogiczne przesłanki leżą u podstaw przyjęcia projektu MNUR.
Podsumowując najważniejsze zmiany procedowane w ramach DNUR należy wskazać, iż proponowane przepisy zawierają regulacje związane z wpływem działalności naukowo-badawczej oraz inwestycyjnej w zakresie ochrony zdrowia na terytorium RP na postępowania refundacyjne. W stosunku do poprzednich wersji przepisów zostało przede wszystkim doprecyzowane w jakim zakresie inwestycje będą wpływały na podejmowanie decyzji refundacyjnych.
Wśród innych przepisów, mających wpływ na cały system ochrony zdrowia, należy wskazać odejście od refundacji opartej na Charakterystyce Produktu Leczniczego w przypadku leków wydawanych w aptece na receptę. Zamiast tego proponowane jest oparcie się na bliżej nieokreślonym „pełnym zakresie wskazań i przeznaczeń”. Pomimo że zmiana ta pojawiła się wyjściowo w projekcie Małej nowelizacji została ona przeniesiona do DNUR.
Podobnie istotne są zmiany związane z zakresem budżetu na refundację, kwestiami obliczania ustawowego paybacku czy ustalaniem limitu finansowania w poszczególnych grupach limitowych. Wszystkie one mają bowiem bezpośredni wpływ na podmioty funkcjonujące na rynku.
Sam projekt DNUR zawiera również nowe, pożądane ze strony systemu ochrony zdrowia, rozwiązania np. dotyczące refundacji leków stosowanych we wskazaniach ultrarzadkich czy szczepionek.
W ramach DNUR proponowane są również przepisy, których wprowadzenie budzi większe wątpliwości np. pozwalające na wszczęcie postępowania dotyczącego wtórnej modyfikacji obowiązujących warunków objęcia refundacją danego produktu. Podobnie nadal proponowane jest uregulowanie w przepisach Prawa farmaceutycznego procedury wczesnego dostępu do nowoczesnych technologii dla polskich pacjentów cierpiących na choroby zagrażające życiu oraz zdrowiu.
Mamy wreszcie przepisy, które porządkują obecny stan prawny. Wśród nich warto wskazać na postulat wprowadzenia odrębnych regulacji dla refundacji leków stosowanych w imporcie 3 równoległym czy w zakresie leków stosowanych w programach lekowych. Innym przykładem są przepisy narzucające obowiązek stosowania urzędowych marż przy okazji eksportu produktów objętych refundacją.
Dodatkowo w ramach MNUR postuluje się wprowadzenie okresu obowiązywania decyzji „do 5 lat” zamiast obecnych 2, 3, 5-letnich okresów. Ponadto proponowane są ograniczenia zakresu danych publikowanych przez NFZ czy wprowadzenie ratunkowego dostępu do terapii. Projekt ten zawiera również szereg zmian doprecyzowujących obecne regulacje prawne.
W związku z ilością zmian oraz dynamiką całego procesu legislacyjnego, który w trakcie ostatnich dni przyspieszył, celem niniejszego alertu nie jest dokonanie ich oceny. Zamiast tego postaramy się przybliżyć Państwu najważniejsze elementy obu projektów.
Z ostateczną oceną procedowanych regulacji należy wstrzymać się do publikacji ostatecznych projektów, a nawet do momentu wejścia w życie ustaw i wytworzenia się odpowiedniej praktyki.
Wpływ na pacjentów:
- możliwość otrzymania leku w „pełnym zakresie wskazań i przeznaczeń”
- zwiększenie dostępu do szczepionek oraz leków stosowanych we wskazaniach ultrarzadkich
- udział organizacji pacjenckich w procesach refundacyjnych (członkostwo w Komisji Ekonomicznej)
Kluczowe zmiany dla hurtowników:
- nakaz stosowania marż urzędowych w eksporcie
- stworzenie odrębnego trybu dla obejmowania refundacją leków z importu równoległego
- dodatkowe kompetencje kontrolne GIF w zakresie wywozu leków
Kluczowe zmiany dla lekarzy:
- swoboda preskrypcyjna dla lekarzy nieograniczona zakresem wskazań z ChPL
- ograniczenia w zakresie możliwości wymierzenia kar za niewłaściwą refundację
Kluczowe zmiany dla producentów:
- ryzyko wszczęcia postępowania dotyczącego zmiany obowiązującej decyzji
- większy wpływ działalności naukowobadawczej i inwestycyjnej na warunki refundacji danego produktu
1. Wydatkowanie środków na refundację
1.1 Zmiany w zakresie budżetu na refundację
- Projektowane przepisy modyfikują zasady tworzenia całkowitego budżetu na refundację. Analogicznie do poprzedniej propozycji – elementami całkowitego budżetu na refundację mają być środki finansowane, które zostały zwrócone do NFZ w związku z ustalonymi w decyzjach refundacyjnych instrumentami dzielenia ryzyka oraz z tytułu ustawowego paybacku. W obecnej wersji nowelizacji doprecyzowano, że środki te w postaci rezerwy, będą włączane do budżetu na refundację w roku następnym po roku za jaki zostały one naliczone. Środki te powinny powiększać budżet refundacyjny.
- W projekcie utrzymano propozycję odejścia od wskazywania przez Ministra Zdrowia w drodze rozporządzenia sposobu podziału środków finansowych, które stanowią wzrost całkowitego budżetu na refundację w danym roku w stosunku do wysokości całkowitego budżetu na refundację w roku poprzednim.
- Do budżetu refundacyjnego nie będą włączane natomiast środki finansowe przekazywane przez Ministra Zdrowia jako dotacja celowa, które mają stanowić budżet na refundacyjny tryb rozwojowy. Środki z tego budżetu będą przeznaczone na pokrycie części kosztów (w wysokości jednej dziesiątej ceny określonej w decyzji o objęciu refundacją i ustaleniu urzędowej ceny zbytu) produktów, które nie mają swojego odpowiednika refundowanego w danym wskazaniu w danej grupie limitowej. W porównaniu do poprzedniej propozycji, z możliwości skorzystania ze środków przekazywanych przez Ministra Zdrowia usunięte zostały leki stosowane we wskazaniach ultrarzadkich.
Ważne! Dotacja celowa na RTR nie będzie włączana do budżetu na refundację. - W projekcie określono maksymalną wysokość kwot przeznaczonych na refundacyjny tryb rozwojowy, które w kolejnych latach działania nowej regulacji mają być systematycznie podwyższane (9,2 mln PLN w 2017 r. do 199,3 mln PLN w 2026 r.).
- Jednocześnie w projekcie wprowadzono mechanizmy umożliwiające Ministrowi Zdrowia zarządzanie wykorzystaniem środków z tego budżetu – z jednej strony poprzez zobowiązanie do zwiększenia budżetu na refundacyjny tryb rozwojowy o środki z tego budżetu, które nie zostały wykorzystane w roku poprzednim, z drugiej zaś poprzez mechanizmy kontroli wysokości wykorzystanych środków w danym roku (przekroczenie po pierwszym półroczu roku kalendarzowego 75% limitu wydatków przewidzianego na ten rok, zobowiązuje Ministra Zdrowia w kolejnym półroczu danego roku do wydania decyzji do wysokości kwoty pozostałej w tym budżecie).
1.2 Zwroty w przypadku przekroczenia budżetu na refundację
- Zgodnie z dotychczasowymi przepisami w przypadku przekroczenia całkowitego budżetu na refundację leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych dostępnych w aptece na receptę, wyznaczane były kwoty przekroczenia dla konkretnych grup limitowych, w ramach których wnioskodawcy byli zobowiązani do zwrotu 50% tego przekroczenia. Mechanizm opierał się na kosztach rocznych, a spod tych regulacji wyłączone były wszystkie produkty, dla których zostały zawarte instrumenty dzielenia ryzyka.
- Projekt przewiduje liczne zmiany dotyczące zarówno mechanizmów ustalania kwot przekroczenia oraz wyliczania kwot zwrotu, jak również leków, które będą wyłączone z obowiązku uczestniczenia w zwrocie.
- Podobnie jak w poprzedniej propozycji zmiany tego przepisu – podstawą do oszacowania kwoty przekroczenia jest pierwotny plan finansowy NFZ na dany rok (nieuwzględniający późniejszych zmian). Natomiast nowością zaproponowaną obecnie jest rozliczanie kwot zwrotu w cyklach kwartalnych (zgodnych z częstotliwością publikacji wykazów refundacyjnych) wraz z referencją do kwot refundacji wydatkowanych na dany produkt w danym kwartale i analogicznym kwartale roku poprzedniego.
- Zaproponowany mechanizm nadal odnosi się jedynie do leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych dostępnych w aptece na receptę. Jednocześnie przesłanką wyłączającą obowiązek zwrotu jest ustalenie w decyzji refundacyjnej instrumentu dzielenia ryzyka uzależniającego wielkości przychodu wnioskodawcy od uzyskiwanych efektów zdrowotnych – czyli w paybacku ustawowym uczestniczyć będą także te produkty, dla których zawarte został inne niż związane z efektem zdrowotnym instrumenty dzielenia ryzyka (w tym także najbardziej popularne w aptekach zobowiązanie do zwrotu części kosztów refundacyjnych do płatnika).
Ważne! Tylko RSS związany z uzyskiwanym efektem zdrowotnym zwalnia z obowiązku zapłaty ustawowego paybacku. - Obowiązek zwrotu nie będzie dotyczył również produktów, które nie wygenerowały wzrostu kwot refundacji na koniec danego kwartału roku rozliczeniowego w porównaniu do kwartału roku poprzedzającego. Wyłączeniu podlegać będą także wnioskodawcy, którzy nie uzyskali przychodów z tytułu refundacji produktu w danej grupie limitowej w danym kwartale roku poprzedzającego – co oznacza, że z mechanizmów wyłączone będą wszystkie produkty dotychczas nierefundowane, a pierwszym okresem naliczania paybacku ustawowego będzie dla nich piąty kwartał obecności w refundacji.
- W pozostałych przypadkach wnioskodawca, który uzyskał decyzję administracyjną o objęciu refundacją, będzie zobowiązany do zwrotu do NFZ kwoty proporcjonalnej do udziału kosztów refundacji w kwocie przekroczenia, w danej grupie limitowej. Przy czym utrzymany został zapis o większym obciążeniu produktów droższych.
- Projektodawca doprecyzowuje, że planowaną kwotą refundacji w danej grupie limitowej dla danego kwartału będzie wartość refundacji dla tej grupy limitowej w analogicznym kwartale roku poprzedniego według planu finansowego NFZ na dzień 31 grudnia powiększona zgodnie ze współczynnikiem wzrostu budżetu.
- W projekcie zaproponowano rozwiązania odnoszące się do sytuacji przeniesienia produktu do odrębnej (nowej lub istniejącej) grupy limitowej albo połączenia grup limitowych.
- Jednocześnie przy wyliczeniu kwot zwrotu zrezygnowano z zastosowania współczynnika „0,5” co oznacza, że wnioskodawcy zobowiązani będą do pokrycia 100% kwoty przekroczenia, a nie jak poprzednio połowy. Kwota przekroczenia w grupie limitowej będzie jak dotychczas pomniejszana współczynnikiem „G”, który odzwierciedla brak przekroczenia planowanej kwoty refundacji w niektórych pozostałych grupach limitowych.
2. Najważniejsze zmiany w zakresie definicji
- Analizowana nowelizacja wprowadza zmiany w zakresie części definicji ustawowych. Całkowitą nowością jest wprowadzenie definicji efektywności klinicznej, którą ustawodawca określa jako bilans korzyści zdrowotnych oraz zdarzeń niepożądanych rozpatrywanej technologii medycznej.
- W przypadku części definicji autorzy doprecyzowali również ich treść w stosunku do poprzednich wersji projektu. W ten sposób zmianie uległa definicja treści programu lekowego, którą stanowić ma zbiór informacji zawierających nazwę międzynarodową leków, warunki ich stosowania w ramach systemu refundacji, a także zasady kwalifikacji, monitorowania i wyłączenia z programu.
Ważne! Zmianie ma ulec definicja odpowiednika leku oraz postulowane jest wprowadzenie definicji opisu programu lekowego. - Definicja programów lekowych ulegnie również zmianie w ramach Małej nowelizacji. Zmiana ma polegać na doprecyzowaniu, że dana substancja czynna nie może być składową kosztową innych świadczeń gwarantowanych w danym wskazaniu oraz dla danej populacji. Oznacza to możliwość obejmowania refundacją tych samych produktów w ramach różnych rodzajów świadczeń oraz dla różnych populacji chorych w danym wskazaniu.
- W analizowanym projekcie pozostawiono również definicje wskazania ultrarzadkiego czy szczepionki. Co istotne, definicje te nie uległy znaczącym zmianom w stosunku do wcześniejszych wersji projektu.
- Wśród doprecyzowanych definicji znalazła się również definicja odpowiednika leku. Dokonano zmiany zapisu referującego do „braku różnic w postaci farmaceutycznej”. Zamiast tego proponuje się odniesienie do leków, które mają taką samą bądź zbliżoną postać farmaceutyczną. W ramach tej definicji dodano również, że odpowiednikami są wyłącznie leki dostępne w tej samej kategorii dostępności refundacyjnej.
- Nowością w tym zakresie jest rozszerzenie definicji odpowiednika leku, w kierunku regulacji prawa farmaceutycznego oraz wskazanie, iż sole, estry, etery, izomery, mieszaniny izomerów, kompleksy lub pochodne substancji czynnej uważa się za taką samą substancję czynną, jeżeli nie różnią się one w sposób znaczący od substancji czynnej swoimi właściwościami w odniesieniu do bezpieczeństwa lub skuteczności.
- Dodatkowo w ramach przedstawionych propozycji zmieniono również definicję przedstawiciela podmiotu odpowiedzialnego, środka spożywczego specjalnego przeznaczenia żywieniowego czy cen detalicznej oraz hurtowej. W tym zakresie mamy raczej do czynienia z niewielkim doprecyzowaniem zakresu tych definicji – zmiany te nie wpływają znacząco na treść przepisów.
3. Odrębności w przypadku postępowań dotyczących wybranych produktów
3.1 Refundacja szczepionek
- Projekt wprowadza definicję szczepionki, rozumianej jako lek będący produktem immunologicznym stosowanym przeciw chorobie zakaźnej w celu sztucznego uodpornienia przeciwko tej chorobie.
- Nowością w stosunku do wcześniejszych wersji projektu jest wskazanie, iż szczepionki stosowane w ramach szczepień zalecanych będą kwalifikowane do odpłatności ryczałtowej. Jednocześnie w zakresie pozostałych przepisów dotyczących szczepionek projektodawca nie precyzuje czy dotyczą one jedynie produktów stosowanych w ramach szczepień zalecanych czy również szczepionek obowiązkowych.
- Minister Zdrowia będzie ustalał grupy limitowe dla szczepionek dostępnych w aptece na receptę. Do danej grupy mają być kwalifikowane szczepionki posiadające ten sam skład jakościowy i ilościowy, albo inny skład jakościowy i ilościowy, ale podobne działanie profilaktyczne i zbliżony mechanizm działania, przy zastosowaniu następujących kryteriów:
- tych samych wskazań lub przeznaczeń, w których są refundowane;
- podobnej skuteczności w zakresie tych samych wskazań.
- Podstawę limitu finansowania w grupie limitowej dla szczepionek dostępnych w aptece na receptę ma nadal wyznaczać najtańsza w grupie szczepionka biorąc pod uwagę pełen cykl szczepień.
3.2 Leki stosowane we wskazaniach ultrarzadkich
- Wskazanie ultrarzadkie zostało zdefiniowane w projekcie jako stan kliniczny występujący nie częściej niż u jednej osoby na 50 000 mieszkańców na terytorium RP lub Unii Europejskiej. Przy czym poprzedni projekt nie precyzował terytorium występowania stanu klinicznego.
- W przypadku leków stosowanych we wskazaniach ultrarzadkich, jeżeli nie posiadają one refundowanego odpowiednika, zostanie wyłączony obowiązek przedstawiania analizy ekonomicznej. Zamiast tej analizy konieczne będzie załączenie do wniosku uzasadnienia ceny.
- Jednocześnie nowością wskazaną w obecnym projekcie jest wyłączenie kryterium wysokości progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość w przypadku leków, dla których zostało dołączone uzasadnienie ceny tj. leków stosowanych we wskazaniach ultrarzadkich. Tym samym wysoki koszt terapii przewyższający wskazany próg nie będzie stanowił przesłanki do odmowy objęcia produktu refundacją.
- Analogicznie, w trakcie negocjacji Komisja Ekonomiczna będzie brała pod uwagę uzasadnienie ceny zamiast progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość w przypadku leku stosowanego we wskazaniu ultrarzadkim nieposiadającego refundowanego odpowiednika.
3.3 Leki z importu równoległego
- W projekcie wyróżniono w ramach odrębnej kategorii produkty pochodzące z importu równoległego. Ich refundacja ma się odbywać na szczególnych zasadach:
- pod warunkiem objęcia refundacją takiego samego leku jak dopuszczony do obrotu w procedurze innej niż import równoległy;
- pod warunkiem ustalenia dla nich urzędowej ceny zbytu nie wyższej niż 85% ceny tego samego leku dopuszczonego do obrotu w innej w procedurze;
- z zapewnieniem weryfikacji zgodności ChPL leku z importu z ChPL takiego samego leku dopuszczonego do obrotu w innej procedurze;
- z uwzględnieniem analizy weryfikacyjnej, stanowiska Rady Przejrzystości i rekomendacji Prezesa AOTMiT przygotowanych w odniesieniu do objętego refundacją takiego samego leku dopuszczonego w innej procedurze.
- Ponadto w Małej nowelizacji zaproponowano wprowadzenie możliwości indywidualnej refundacji na podstawie zgody Ministra Zdrowie dla leku sprowadzanego z zagranicy niedostępnego na terytorium RP. Obecnie przepisy dopuszczają jedynie tę procedurę dla leków nieposiadających pozwolenia na dopuszczenie do obrotu.
4. Obejmowanie produktów refundacją
4.1 Leki złożone
- Nowością dotychczas nieuregulowaną w ustawie refundacyjnej jest propozycja nowego podejścia do leków złożonych. Zamiast dotychczasowej zasady 1+1 = 1 w opublikowanym projekcie zaproponowano uwzględnienie obu substancji czynnych w leku – zarówno na etapie obejmowania leku złożonego refundacją, jak również w późniejszych wyliczeniach cen, limitów oraz dopłaty pacjentów, a także rygorów progowych zakupu tych produktów przez świadczeniodawców w ramach świadczeń gwarantowanych. Regulacja odnosi się zarówno do produktów złożonych stosowanych w lecznictwie zamkniętym, jak również leków dostępnych w aptece na receptę.
- Jednocześnie dla leku złożonego zawierającego dwie lub więcej substancji, które objęte są refundacją, w przypadku braku różnic w efektywności klinicznej – wprowadzono obowiązek złożenia we wniosku o objęcie refundacją analizy wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych.
4.2 Rezygnacja z obowiązku stosowania ChPL w refundacji
- Jednym z kluczowych elementów obecnego projektu jest pojawiająca się we wcześniejszych projektach zmiany ustawy o refundacji propozycja odejścia od oparcia wskazania refundacyjnego o wskazanie określone zgodnie z treścią ChPL. Zamiast tego proponowane są postanowienia wskazujące na refundację „w pełnym zakresie wskazań i przeznaczeń”.
Ważne! Leki dostępne w aptece na receptę w całym zakresie zarejestrowanych wskazań będą dostępne „w pełnym zakresie wskazań i przeznaczeń”. - Niestety ustawodawca nie definiuje co oznacza słowo „pełny”. W poprzednich propozycjach wskazywano, iż chodzi o całość wskazań i przeznaczeń ustalonych na podstawie aktualnej wiedzy medycznej1. Niestety nie wynika to wprost z treści proponowanego przepisu.
- Należy jednocześnie pamiętać, że procedowana zmiana nie powoduje, że wszystkie refundowane dotychczas leki będę dostępne w w/w zakresie wskazań. Ograniczenia do konkretnych wskazań (stanów klinicznych) nadal będą obowiązywały dla leków refundowanych w aptece w danym wskazaniu oraz w chemioterapii czy programach lekowych.
- Przepis nie precyzuje do jakich wskazań oraz przeznaczeń referuje projektodawca. Nie zostało również wskazane w jaki sposób lekarz ma udowodnić prawidłowość preskrypcji w razie kontroli NFZ.
4.3 Nowy sposób ustalania treści programów lekowych
- Do projektu włączono również rozwiązanie problemu braku elastyczności programów lekowych. Przedmiotowa propozycja nie uległa zmianie w stosunku do wcześniejszych wersji projektu.
- Zamiast opisu całego programu lekowego, stanowiącego wspólny załącznik kilku decyzji, w decyzjach refundacyjnych osobno ustalane mają być: o nazwa programu lekowego, do jakiego kwalifikowany zostanie określony lek, o warunki jego stosowania w programie lekowym w formie załącznika do decyzji.
- Opis programu lekowego nie będzie już zatem załącznikiem do poszczególnych decyzji. Zamiast tego stanie się zbiorem informacji z konkretnych decyzji administracyjnych (z załączników określających warunki stosowania) oraz być może innych warunków dla wszystkich leków, które mają być finansowane w danym wskazaniu, w całości zawartym w treści obwieszczenia refundacyjnego.
Ważne! Obecne brzmienie przepisów nie wskazuje, jakie warunki objęcia refundacją w ramach programu lekowego zostaną zawarte w decyzjach refundacyjnych. - Obecne brzmienie projektu nie wskazuje jakie elementy programu pozostaną poza treścią decyzji refundacyjnej oraz będą mogły zostać zmienione w trakcie obowiązywania tej decyzji bez konieczności uzyskania zgody wnioskodawcy. Nie zostało bowiem zdefiniowane pojęcie warunków objęcia refundacją, które zgodnie z wcześniejszymi wyjaśnieniami powinny zostać uwzględnione w treści decyzji refundacyjnej.
- Odrębnie uregulowano, że opis programu lekowego może przewidywać powołanie przez Prezesa Funduszu zespołu koordynacyjnego dla danego programu lekowego, do którego zadań należeć będzie kwalifikacja świadczeniobiorców do programu lekowego, ocena skuteczności terapii w trakcie trwania programu lekowego oraz rozstrzyganie o wyłączeniu świadczeniobiorców z programu lekowego.
4.4 Nowe kryteria refundacyjne
- Autorzy projektu proponują również zmiany w zakresie kryteriów refundacyjnych branych od uwagę przy wydawaniu decyzji refundacyjnej. Tak jak w poprzednich propozycjach nowelizacji ustawy, nowym kryterium jest opinia ministra do spraw gospodarki.
- Przesłanką, która pojawia się pierwszy raz, jest natomiast miara rozrzutu i niepewność oszacowań wyników analizy klinicznej, analizy ekonomicznej, analizy wpływu na budżet podmiotu zobowiązanego do finansowania świadczeń ze środków publicznych.
- Zmianie ulega również kryterium dotyczące wysokości progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość. Obok możliwości określania progu opłacalności na podstawie analizy użyteczności kosztów oraz analizy efektywności kosztów w projekcie dopuszczona została także analiza minimalizacji kosztów. Jednocześnie w tej wersji nowelizacji pojawia się nowość związana z wyłączeniem uwzględniania przedmiotowego kryterium dla leków stosowanych w chorobach ultrarzadkich.
4.5 Nowe regulacje w zakresie instrumentów dzielenia ryzyka
- W obecnej wersji projektu utrzymano propozycję rozszerzenia zakresu instrumentów dzielenia ryzyka. Do wskazanych dotychczas możliwości ustalenia innych warunków refundacji, mających wpływ na zwiększenie dostępności do świadczeń gwarantowanych dodano szereg nowych elementów.
- Istotne jest potwierdzenie również w dużej nowelizacji ustawy refundacyjnej, iż w przypadku kiedy część kosztu produktu objętego refundacją ma być pokrywana z budżetu na refundacyjny tryb rozwojowy, w decyzji obligatoryjnie zawiera się instrument dzielenia ryzyka polegający na wskazaniu kwoty wydatków po przekroczeniu której wnioskodawca jest obowiązany do zwrotu całej kwoty przekroczenia.
Ważne! Dla produktów finansowanych w ramach budżetu na RTR – proponuje się wprowadzić nakaz zawarcia instrumentu w postaci zwrotu kwoty przekroczenia powyżej ustalonego poziomu. - Dodatkowo rozszerzono możliwość zawierania instrumentów dzielenia ryzyka w decyzji wydanej w przedmiocie ustalenia urzędowej ceny zbytu dla produktów finansowanych w ramach innych świadczeń gwarantowanych. Taka możliwość nie wynika bowiem z obecnej treści przepisów, co powodowało w przeszłości wątpliwości dotyczące ich stosowania.
4.6 Decyzje off-label
- Możliwość stosowania refundacji off-label do wszystkich kolejnych odpowiedników, dzięki wykreśleniu warunku braku innych możliwych do zastosowania w danym stanie klinicznym procedur medycznych finansowanych ze środków publicznych.
4.7 Warunki dalszej refundacji
- Nowelizacja nadal wprowadza zmianę w przepisie regulującym warunki wydawania decyzji refundacyjnych dla produktów objętych wcześniej refundacją.
- Zgodnie z obecnym brzmieniem, wnioskodawca nie może oczekiwać ustalenia dla takiego produktu urzędowej ceny zbytu wyższej niż dotychczas ustalona. Proponowane brzmienie przepisów wskazuje, że sumaryczna ocena urzędowej ceny zbytu oraz mechanizmu dzielenia ryzyka nie może być mniej korzystna dla płatnika publicznego niż obowiązująca w dniu złożenia kolejnego wniosku. W stosunku do poprzedniej propozycji dookreślone zatem zostało o czyjej korzyści mowa.
Ważne! W przypadku uchwalenia proponowanych przepisów, warunki wejścia w życie kolejnej decyzji refundacyjnej nie będą mogły być mniej korzystne dla płatnika publicznego. - W przypadku prostych instrumentów dzielenia ryzyka przewidujących wyłącznie świadczenia na rzecz NFZ, ocena wynikających z nich korzyści nie powinna nastręczać problemów. Mogą one jednak wystąpić w przypadku bardziej złożonych instrumentów lub w przypadku chęci zastąpienia jednego instrumentu innym – opartym na odmiennym mechanizmie działania.
- Przedmiotowy przepis wymusza ostrożność w przypadku zawierania instrumentów dzielenia ryzyka związanych z tzw. capem oraz zobowiązaniem wnioskodawcy do zwrotu 100% kwoty refundacji. Ustalony w takim instrumencie poziom finansowania przez NFZ będzie obowiązkowo punktem wyjścia w przypadku kontynuacji refundacji.
4.8 Opinia ministra właściwego do spraw gospodarki
- Przedmiotowy projekt przewiduje kompetencję ministra właściwego do spraw gospodarki – obecnie Ministra Rozwoju – do wydawania opinii o działalności gospodarczej, w tym działalności naukowo-badawczej i inwestycyjnej w zakresie związanym z ochroną zdrowia na terytorium RP prowadzonej przez podmiot ubiegający się o refundację produktu.
- W przeciwieństwie do poprzedniego projektu, w obecnym zostały precyzyjnie wskazane zarówno elementy brane pod uwagę przez Ministra Rozwoju przy wydawaniu opinii, jak również skutki uzyskania określonej kategorii. W poprzednich wersjach projektu mieliśmy wyłącznie odniesienie do elementów, które powinny być wskazane we wniosku, a które jak się wydawało powinny być brane pod uwagę przy wydawaniu opinii.
- Obecnie doprecyzowany został również zakres przedstawianych informacji. Co istotne, przyjęty został jeden z postulatów, nad którym wcześniej pracowaliśmy – wnioskodawca przedstawia informacje dotyczące prowadzonej działalności gospodarczej w odniesieniu do grupy kapitałowej, do której należy, w ujęciu rocznym za okres odpowiednio ostatnich trzech lub pięciu lat obrachunkowych poprzedzających datę złożenia wniosku.
- Pod rygorem odpowiedzialności karnej za składanie fałszywych zeznań będą przekazywane informacje dotyczące następujących obszarów:
|
INNOWACJA |
nakłady na działalność badawczo-rozwojową, wewnętrzne i zewnętrzne |
|
łączne nakłady poniesione na badania podstawowe i przedkliniczne (w tym badania bioinformatyczne) prowadzone na terytorium RP |
|
|
łączne nakłady poniesione na badania kliniczne prowadzone na terenie RP |
|
|
koszty działalności operacyjnej – ogółem |
|
|
PRODUKCJA |
suma kwot amortyzacji majątku produkcyjnego na syntezę chemiczną |
|
suma kwot amortyzacji majątku produkcyjnego na technologię nuklearną |
|
|
suma kwot amortyzacji majątku produkcyjnego na biotechnologię |
|
|
suma kwot amortyzacji majątku produkcyjnego na inne technologie medyczne |
|
|
przychody netto ze sprzedaży towarów i usług |
|
|
INWESTYCJE |
inwestycje w wyposażenie laboratoriów badawczo-rozwojowych |
|
łączne nakłady na inwestycje w urządzenia techniczne (np. do analizy jakości) i maszyny produkcyjne |
|
|
łączne nakłady na inwestycje bezpośrednio poniesione na majątek na terytorium RP |
|
|
łączne nakłady na inwestycje bezpośrednio poniesione na start-up'y działające na terenie RP będące podatnikiem na terytorium RP |
|
|
ZATRUDNIENIE |
łączna wartość odprowadzonych zaliczek na podatek dochodowy od osób fizycznych za pracownika (PIT-4) |
|
łączna liczba równoważników etatów osób aktualnie zatrudnionych w działalności badawczorozwojowej |
|
|
łączna liczba równoważników etatów osób aktualnie zatrudnionych przy działalności produkcyjnej i czynnościach kontrolnych |
|
|
PODATKI |
łączna wartość odprowadzonego podatku od osób prawnych (CIT) |
|
EKSPORT |
wartość eksportu towarów |
|
wartość eksportu usług |
|
|
wartość importu półproduktów i materiałów |
- W ramach drugiego z procedowanych wcześniej postulatów w nowym projekcie pojawiło się wskazanie, że dane przekazywane w ramach wniosku o wydanie opinii oraz opinia będą stanowiły tajemnicę przedsiębiorcy. Do publicznej wiadomości będzie przekazywana wyłącznie informacja o wydanych opiniach wraz z podaniem podmiotu wnioskującego oraz przyznanej mu kategorii, zawartej w opinii.
Ważne! Informacje prezentowane przez wnioskodawcę będą dotyczyły całej grupy kapitałowej. - W celu uwzględnienia opinii w postępowaniu refundacyjnym konieczne będzie przekazanie zestawienia wskazanych informacji Ministrowi Zdrowia do dnia 30 czerwca każdego roku. W przypadku dołączenia opinii do wniosku refundacyjnego i nieprzekazania zestawienia informacji w terminie, opinia nie będzie uwzględniania w roku następnym po roku niedopełnienia obowiązku.
- Podmiot wnioskujący będzie mógł wystąpić z wnioskiem o wydanie opinii uwzględniającej także przyszłe inwestycje. Opinia taka będzie miała charakter poglądowy i nie będzie mogła stanowić załącznika do wniosku refundacyjnego. W tym miejscu powstanie pytanie w jaki sposób opinia ta będzie wpływała na postępowania refundacyjne.
- Minister Rozwoju będzie wydawał decyzję w terminie 30 dni od dnia złożenia wniosku. W przypadku, gdy wniosek nie zawiera wymaganych informacji Minister Rozwoju będzie niezwłocznie informował podmiot wnioskujący o konieczności uzupełnienia w terminie nie dłuższym niż 14 dni. Termin wydania decyzji ulegnie zawieszeniu do dnia uzupełnienia wniosku. Nieuzupełnienie wniosku w terminie będzie skutkowało pozostawieniem wniosku bez rozpatrzenia.
- Nowością jest wskazanie w przepisach przejściowych, że Minister Zdrowia w terminie 30 dni od dnia wejścia w życie ustawy będzie wzywał wnioskodawców, których produkty będą objęte refundacją w dniu wejścia w życie ustawy do przedstawienia zestawienia informacji dotyczących prowadzonej działalności.
- Opłata za złożenie i uzupełnienie wniosku wyniesie do 5 000 zł i zostanie określona przez Ministra Rozwoju w porozumieniu z Ministrem Zdrowia w drodze rozporządzenia. Analogicznie zostanie ustalony sposób przedstawienia informacji, wagi przypisane do poszczególnych informacji, algorytm wyliczenia całkowitego wyniku punktowego, na podstawie którego następuje przyznanie określonej kategorii oraz zakres punktowy poszczególnych kategorii.
- Poniższa tabela przedstawia korzyści uzyskiwane przez wnioskodawców posiadających opinię o wpływie działalności na gospodarkę o określonej kategorii:
|
KATEGORIA A |
KATEGORIA B |
KATEGORIA C |
KATEGORIA D |
|
wartość progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość będzie powiększona o 5% |
wartość progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość będzie powiększona o 10% |
wartość progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość będzie powiększona o 15% |
wartość progu kosztu uzyskania dodatkowego roku życia skorygowanego o jakość będzie powiększona o 20% |
|
UCZ pierwszego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym nie może być wyższa niż 80% UCZ odpowiednika wyznaczającego podstawę limitu finansowania |
UCZ pierwszego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym nie może być wyższa niż 85% UCZ odpowiednika wyznaczającego podstawę limitu finansowania |
UCZ pierwszego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym nie może być wyższa niż 90% UCZ odpowiednika wyznaczającego podstawę limitu finansowania |
UCZ pierwszego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym nie może być wyższa niż 95% UCZ odpowiednika wyznaczającego podstawę limitu finansowania |
|
UCZ kolejnego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym, może być wyższa o 5% niż UCZ odpowiednika wyznaczającego podstawę limitu albo najtańszego odpowiednika o ile podstawę limitu w danej grupie limitowej wyznacza lek z inną substancją czynną |
UCZ kolejnego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym, może być wyższa o 15% niż UCZ odpowiednika wyznaczającego podstawę limitu albo najtańszego odpowiednika o ile podstawę limitu w danej grupie limitowej wyznacza lek z inną substancją czynną |
UCZ kolejnego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym, może być wyższa o 20% niż UCZ odpowiednika wyznaczającego podstawę limitu albo najtańszego odpowiednika o ile podstawę limitu w danej grupie limitowej wyznacza lek z inną substancją czynną |
UCZ kolejnego odpowiednika z uwzględnieniem liczby DDD w opakowaniu jednostkowym, może być wyższa o 25% niż UCZ odpowiednika wyznaczającego podstawę limitu albo najtańszego odpowiednika o ile podstawę limitu w danej grupie limitowej wyznacza lek z inną substancją czynną |
4.9 Grupy limitowe i limity finansowania
- W projekcie powtórzono propozycję doszczegółowienia kwestii pojawiających się wraz z objęciem refundacją kolejnych pierwszych odpowiedników w ramach jednej grupy limitowej – podstawę limitu wyznaczać będzie każdorazowo pierwszy odpowiednik, którego cena hurtowa brutto za 1 DDD pozostaje na najniższym poziomie.
- Analogicznie, jak przy poprzednim projekcie nowelizacji – obecnie także rozszerzono katalog przypadków, w których dla ustalenia podstawy limitu i wyliczenia limitów finansowania brana pod uwagę może być najczęściej stosowana dobowo dawka leku (PDD) zamiast DDD. Dotychczas było to możliwe jedynie w przypadku, gdy DDD było niższe od PDD oraz, jak można przyjąć, w razie braku ustalenia DDD dla danej substancji czynnej. Zgodnie z proponowaną zmianą PDD będzie mogło być brane pod uwagę również wtedy, gdy będzie niższe od wartości opartej na DDD.
- W ramach doprecyzowania kwestii funkcjonowania pierwszych odpowiedników Minister Zdrowia postuluje również dookreślenie, że obniżenie ceny do poziomu 75% ceny zbytu jedynego odpowiednika nie będzie dotyczyło produktów posiadających tę samą nazwą handlową. W ten sposób z regulacji dotyczących pierwszego odpowiednika zostaną wyłączone kolejne prezentacje refundowanego wcześniej leku oraz leki z importu równoległego.
4.10 Tworzenie nowych grup limitowych
- W obecnej wersji projektu Małej nowelizacji zrezygnowano z wprowadzenia dodatkowej przesłanki wyodrębnienia nowej grupy limitowej – w pierwotnym zamierzeniu Minister Zdrowia miał mieć możliwość utworzenia odrębnej grupy dla danego produktu w przypadku ustalenia w decyzji instrumentów dzielenia ryzyka, uwzględniając wpływ na budżet płatnika publicznego oraz uzyskanie dodatkowych efektów zdrowotnych.
4.11 Negocjacje z Komisją Ekonomiczną
- Najważniejszą zmianą w zakresie proponowanych przepisów jest zmiana w zakresie składu Komisji Ekonomicznej poprzez dodanie przedstawiciela organizacji pacjenckich. Co istotne, projektowane przepisy nie wskazują w jaki sposób ten członek Komisji Ekonomicznej będzie wybierany.
Ważne! Oprócz proponowanych wcześniej zmian w zakresie kryteriów uwzględnianych w negocjacjach z KE oraz wymagań dotyczących jej członków, nowa wersja projektu proponuje wprowadzenie do składu KE przedstawiciela organizacji pacjenckich. - Projekt przewiduje również nowe kryteria uwzględniane w trakcie negocjacji z Komisją Ekonomiczną. Kryteria te obejmą opinię ministra właściwego do spraw gospodarki, konkurencyjność cenową, wysokość dopłat świadczeniobiorców oraz uzasadnienie ceny dla leków stosowanych we wskazaniach ultrarzadkich.
- Nowością w stosunku do wcześniejszej wersji przepisów jest wyłączenie kryterium progu opłacalności w stosunku do leków stosowanych we wskazaniach ultrarzadkich.
- Ponadto zaproponowano dodatkowy wymóg dla członków Komisji Ekonomicznej – obowiązek posiadania wiedzy w zakresie medycyny opartej na faktach, oceny technologii medycznej, budowy instrumentów dzielenia ryzyka oraz sposobu kształtowania cen produktów obejmowanych refundacją. Dodatkowo wymagana ma być znajomość języka angielskiego na poziomie B2 potwierdzona certyfikatem.
4.12 Nowe elementy decyzji refundacyjnej
- Oprócz wskazywanego wcześniej dookreślenia w treści decyzji refundacyjnej informacji związanych z refundacją danego leku w programie lekowym wprowadzone zostały również inne elementy. W tym zakresie autorzy projektu proponują wprowadzenie informacji dotyczących:
- wielkości zadeklarowanych dostaw, jeżeli dotyczy,
- refundowanego wskazania klinicznego,
- wartości w jakiej dany produkt jest finansowany z budżetu na refundacyjny tryb rozwojowy.
5. Funkcjonowanie produktu w systemie refundacji
5.1 Zmiana decyzji refundacyjnych z urzędu
- W najnowszej wersji projektu zachowano przepisy przewidujące możliwość wszczęcia z urzędu postępowania w sprawie zmiany decyzji refundacyjnych. Jak wynikało z uzasadnienia projektu, miało być to związane z faktem, że decyzje te będą wydawane na coraz dłuższe okresy, co ma czynić zasadnym wprowadzenie rozwiązań pozwalających na dokonanie ponownej analizy istotnych okoliczności związanych z refundacją danego leku.
- Rozwiązanie to budzi wątpliwości, pozostawiając bez odpowiedzi pytania o szczegółowy przebieg oraz ostateczny cel takiego postępowania. Decyzja o objęciu produktu refundacją wraz z uzyskaniem przez nią waloru ostateczności nie może być bowiem obecnie zmieniona ani uchylona bez zgody wnioskodawcy.
- Projektowana zmiana – choć nie wprost – zdaje się zmierzać w kierunku ustanowienia wyłomu od tej zasady. Co istotne, uzasadnieniem dla wprowadzanego trybu zmiany decyzji refundacyjnych nie może być wydłużający się okres ich obowiązywania – służyć ma on właśnie zwiększeniu stabilności rynku.
Ważne! Proponowane przepisy wprowadzają możliwość wszczęcia postępowania w przedmiocie zmiany obowiązującej decyzji. Nie zostało jednocześnie wskazane czy w takim postępowaniu może zostać wydana decyzja wbrew woli danego wnioskodawcy. - W projekcie przewidziano możliwość przeprowadzenia negocjacji z wnioskodawcą, zasięgnięcia opinii AOTMiT, opinii konsultanta krajowego oraz opinii ministra właściwego do spraw gospodarki – trudno jednak ustalić, jaki ma być ostateczny cel tego postępowania.
- Przyjęcie, że mogłoby ono doprowadzić do zmiany warunków refundacji bez zgody wnioskodawcy stoi w sprzeczności z zasadą praw nabytych. Postępowanie takie mogłoby stanowić płaszczyznę dla wypracowania nowego konsensusu między Ministrem Zdrowia a wnioskodawcą, konieczne jest jednak dookreślenie jego reguł z poszanowaniem praw adresatów decyzji refundacyjnych.
5.2 Okres obowiązywania decyzji refundacyjnych
- W Małej nowelizacji postulowane jest wprowadzenie okresu obowiązywania decyzji do 5 lat. Na długość okresu mają mieć wpływ:
- wynik negocjacji z Komisją Ekonomiczną,
- informacja o wszczętych postępowaniach w sprawie dopuszczenia do obrotu na terenie RP oraz pozostałych państw członkowskich UE lub EFTA,
- okres wyłączności rynkowej,
- informacja o prowadzonych badaniach klinicznych we wnioskowanym wskazaniu.
- Nowelizacja grudniowa wprowadzała możliwość wydania decyzji z okresem obowiązywania do 5 lat, obok dotychczasowych rozwiązań. Obecna propozycja likwiduje możliwość wydawania decyzji na okres 2, 3 i 5 lat. Wedle ostatniego projektu zmian jedyną możliwością jest wydanie decyzji na okres do 5 lat.
5.3 Marże urzędowe w eksporcie
- Analizowany projekt ma w swoim zamierzeniu dookreślić zasady obrotu produktami refundowanymi przeznaczonymi na eksport. W okresie obowiązywania ustawy o refundacji pojawiały się rozbieżności co do tego, czy hurtownia sprzedająca produkt refundowany za granicę zobowiązana jest do stosowania marż urzędowych.
- Projektowany przepis wskazuje wprost, że marże urzędowe znajdą zastosowanie w stosunku do leków, które zostały przeznaczone na wywóz poza granicę terytorium RP.
Ważne! Proponowane przepisy wprowadzają obowiązek stosowania urzędowych ceny oraz marż nie tylko na terytorium RP, ale również w ramach eksportu. - Dodatkowo zaproponowano zwiększenie nadzoru nad lekami podlegającymi wywozowi. W ramach zmiany Prawa farmaceutycznego, która jest częścią analizowanego projektu, wprowadzono również możliwość zwrócenia się inspekcji farmaceutycznej do organów podatkowych oraz Ministra Finansów o udostępnienie informacji w celu:
- ustalenia czy przedsiębiorca prowadzący hurtownię nie dokonał wywozu produktów leczniczych poza teren RP wbrew obowiązującymi przepisom,
- określenia wielkości obrotu w/w produktami oraz wymierzenie kary.
5.4 Publikacja informacji o złożonych wnioskach
- Projekt przewiduje również publikację informacji o złożonych wnioskach refundacyjnych w BIP Ministra Zdrowia oraz na stronie Ministerstwa Zdrowia. Wśród udostępnianych informacji będzie wskazana nazwa produktu, wnioskodawca, wnioskowana kategoria dostępności, wnioskowane wskazanie, warunki stosowania (w przypadku produktów, które mają być refundowane w ramach programów lekowych) oraz etap rozpatrywania wniosku.
- Jednocześnie będą publikowane uwagi zgłaszane do wniosków włączane do akt danej sprawy administracyjnej. Uwagi będą mogły być zgłaszane przez konsultantów z danej dziedziny medycyny oraz stowarzyszenia i fundacje, których cele statutowe są związane z przedmiotem wniosku.
5.5 Obwieszczenie refundacyjne
- W projekcie została zmieniona częstotliwość wydawania obwieszczeń refundacyjnych – mają być wydawane co 3 miesiące, a nie jak obecnie co 2.
- Zmiana częstotliwości publikacji wykazów refundacyjnych znajdzie odzwierciedlenie w algorytmie kalkulacji limitu finansowania. Podstawa limitu w danej grupie limitowej będzie wyznaczana w oparciu o dane z ostatniego z trzech miesięcy działania dwóch edycji poprzedzających publikację wykazu refundacyjnego, a nie jak obecnie z pierwszego miesiąca. Zatem dane o obrocie będą miały bardziej stabilny charakter.
Ważne! Obwieszczenia refundacyjne będą wydawane co 3 miesiące – zamiast 6 obwieszczeń w skali roku będą wydawane 4. - Ponadto przewidziany został przepis przejściowy, mający zastosowanie dla decyzji refundacyjnych, których okres obowiązywania upłynie po wejściu w życie nowelizacji, ale przed dniem ogłoszenia kolejnego obwieszczenia. W takim przypadku okres obowiązywania decyzji ulega przedłużeniu do dnia wydania tego obwieszczenia.
5.6 Publikowanie danych dotyczących refundacji
- Wśród projektowanych zmian pojawia się również postulat ograniczenia informacji publikowanych przez NFZ dotyczących refundacji. Zgodnie ze zmianami wskazanymi w Małej nowelizacji do publicznej wiadomości będzie podawana jedynie informacja o wielkości kwoty refundacji poszczególnych produktów, ale bez wskazania liczby zrefundowanych opakowań. Wskazana zmiana ma na celu zapewnienie poufności instrumentów dzielenia ryzyka – obecnie na podstawie danych NFZ możliwe jest bowiem wyliczenie wielu algorytmów, a tym samym naruszana jest tajemnica przedsiębiorstwa adresata decyzji.
5.7 Program indywidualnego stosowania produktu leczniczego
- Projekt przewiduje również zmianę w Prawie farmaceutycznym umożliwiającą, w określonych przypadkach, zastosowanie leków jeszcze niezarejestrowanych – dla których został złożony wniosek o dopuszczenie do obrotu lub które są w trakcie badań klinicznych.
Ważne! Proponowane przepisy regulują wprost wprowadzenie tzw. Early Access Program. Dotychczas nasi klienci prowadzili te działania na podstawie wypracowanej przez nas praktyki. - Leki te będą dostępne dla określonej grupy pacjentów cierpiących na przewlekłą lub poważną, wycieńczającą lub zagrażającą życiu chorobę, jeżeli na terytorium RP nie jest możliwa inna, skuteczna technologia medyczna.
- Procedura zastosowania wskazanych leków wymaga zgody Ministra Zdrowia na wniosek podmiotu odpowiedzialnego lub sponsora badania klinicznego, który jest rozpatrywany w terminie nie dłuższym niż 30 dni (w odróżnieniu do 90-dniowego terminu proponowanego w poprzednim projekcie). Wniosek obejmuje m.in. zobowiązanie do zapewnienia dostępności i wskazanie jego realizacji oraz określenie sposobu finansowania, należy również załączyć informacje związane z zastosowaniem produktu leczniczego. Minister Zdrowia może zasięgnąć opinii Europejskiej Agencji Leków, jak również konsultanta z danej dziedziny medycyny.
- W przypadku uzyskania zgody możliwe jest również sprowadzenie danego leku z zagranicy, które należy zgłosić Ministrowi Zdrowia. Wśród obowiązków wnioskodawcy, który uzyskał zgodę, znajduje się monitorowanie bezpieczeństwa produktu leczniczego.
- Powyższa zmiana implementuje wskazaną w prawie unijnym instytucję wczesnego udostępniania produktu leczniczego (ang. Early Access Program), zapewniając pacjentom dostęp do niezbędnej terapii. Postulat sformalizowania procedury dostępu do programu był niejednokrotnie podnoszony przez pacjentów, jak również firmy farmaceutyczne.
5.8 Ratunkowy dostęp do technologii lekowych
- Obejmuje przypadki uzasadnionej i wynikającej ze wskazań aktualnej wiedzy medycznej potrzeby zastosowania leku przy wyczerpaniu wszystkich możliwych do zastosowania w tym wskazaniu refundowanych technologii medycznych, jeżeli jest to niezbędne dla ratowania życia i zdrowia pacjentów.
- Dotyczy leków, które są dopuszczone do obrotu lub pozostają w obrocie oraz są dostępne na rynku, ale nie są finansowane ze środków publicznych w danym wskazaniu.
- Minister Zdrowia, na wniosek świadczeniodawcy, będzie wydawał indywidualne zgody na pokrycie kosztów leku na okres nie dłuższy niż trzymiesięczna terapia albo trzy cykle leczenia. Decyzji nadawany jest rygor natychmiastowej wykonalności.
- Minister Zdrowia może wydać kolejną zgodę w ramach kontynuacji leczenia w przypadku potwierdzenia skuteczności terapii przez lekarza specjalistę.
- Dodatkowo Minister Zdrowia zleca AOTMiT przygotowanie opinii w zakresie zasadności finansowania ze środków publicznych wnioskowanego leku w danym wskazaniu, jeżeli:
- koszt wnioskowanej terapii w ujęciu miesięcznym lub jeden cykl leczenia przekraczają jedną czwartą PKB na jednego mieszkańca,
- uprzednio została wydana co najmniej jedna zgoda dla danego produktu w sprawie wniosku innego świadczeniobiorcy.
- Obecnie ustawodawca zrezygnował z ustanowienia maksymalnej marży hurtowej dla leków finansowanych w ramach ratunkowego dostępu do technologii lekowych. W projekcie grudniowym marża była przewidziana na maksymalnym poziomie 5%, natomiast w projekcie pierwotnym wynosiła ona 10%, ale nie więcej niż 200 zł.
5.9 Kary administracyjne
- Projekt przewiduje dodatkowe kary związane z kontrolami prowadzonymi przez NFZ. Fundusz ma zostać wyposażony w kompetencję do nakładania kar na podmiot prowadzący aptekę, który uniemożliwia czynności kontrolne, nie wykonuje w terminie zaleceń pokontrolnych albo naruszył obowiązki związane z realizacja i rozliczaniem recept w tym w zakresie stosowania limitów, cen, odpłatności i dopłaty świadczeniobiorcy w wysokości określonej w obwieszczeniu oraz stosowania marży detalicznych.
- Kary nakładane przez NFZ będą mogły osiągnąć równowartość kwoty refundacji za okres objęty kontrolą.
- W najnowszej wersji projektu zrezygnowano z przeniesienia kompetencji do nakładania kar administracyjnych z tytułu naruszenia cen i marż urzędowych na organy Państwowej Inspekcji Farmaceutycznej.
- Zachowano natomiast postanowienia dotyczące kar pieniężnych za niedotrzymanie postanowień w zakresie instrumentów dzielenia ryzyka. Ich wysokość wynosić ma do dwukrotności wartości wynikającej z instrumentu, a w przypadku gdy nie jest możliwe jej wyliczenie – do wysokości wartości rocznego obrotu produktami tego wnioskodawcy.
- Co istotne nie została wyjaśniona „wartość wynikająca z instrumentu”. Jest to pojęcie nieostre, które nie zostało doprecyzowane w projekcie. W zależności od przyjętej interpretacji – możliwość wymierzenia kary do wysokości rocznego obrotu danego wnioskodawcy, przy literalnym brzmieniu przepisu uwzględniającym wszystkie produkty, nie tylko te, których dotyczy instrument. Wprowadzono również możliwość miarkowania powyższej kary – dotychczas była określona sztywno na podstawie dwukrotności strat ponoszonych przez Narodowy Fundusz Zdrowia.
- Zmianie ulec ma charakter sankcji z tytułu niedotrzymania zobowiązania do zapewnienia wielkości i ciągłości dostaw. Dotychczas wiązało się ono z obowiązkiem zwrotu określonej kwoty do NFZ.
- Dodana została natomiast nowa kara administracyjna wymierzana w wysokości kwoty stanowiącej iloczyn liczby niedostarczonych w danym roku jednostkowych opakowań danego produktu oraz ich urzędowej ceny zbytu netto. Karę tę będzie nakładał Główny Inspektor Farmaceutyczny.
5.10 Nowe zasady rozliczania refundacji przez apteki
- Projekt przewiduje zastąpienie kontraktów zawieranych z NFZ stosunkiem prawnym wynikającym wprost z obowiązujących przepisów. Zgodnie z projektowanymi rozwiązaniami, apteka zobowiązana będzie jedynie przekazać do NFZ:
- kopię zezwolenia na prowadzenie apteki,
- imię i nazwisko osoby będącej kierownikiem apteki wraz z kopią dokumentów uprawniających do pełnienia tej funkcji,
- aktualną ewidencję osób zatrudnionych w aptece wraz z numerami dokumentów uprawniających do wykonywania zawodu,
- numer rachunku bankowego podmiotu prowadzącego aptekę.
- Podstawę do rozliczenia recept na leki refundowane ma być przekazanie do właściwego oddziału wojewódzkiego Funduszu uzgodnionego zestawienia zbiorczego w formie pisemnej lub elektronicznej.
6. Przepisy przejściowe i zmiany w innych aktach
- Projekt przewiduje kilka zmian w zakresie reklamy leków:
- Zmienia wymogi, jakie spełniać mają katalogi handlowe i listy cenowe. W razie wskazania na nich produktów, dla których wydano decyzję refundacyjną lub decyzję o ustaleniu urzędowej ceny zbytu, na liście cenowej wystarczyło zamieszczenie ceny urzędowej detalicznej.
- Zgodnie z projektem obowiązkowo będą musiały znaleźć się nań również informacje o ich kategorii dostępności refundacyjnej, zakresie wskazań objętych refundacją, poziomie odpłatności, urzędowej cenie zbytu oraz aktualnej wysokości dopłaty świadczeniobiorcy.
- Te same informacje (o kategorii dostępności refundacyjnej, zakresie wskazań objętych refundacją, poziomie odpłatności, urzędowej cenie zbytu oraz aktualnej wysokości dopłaty świadczeniobiorcy) zawierać będzie musiała reklama skierowana do osób uprawnionych do wystawiania recept lub osób prowadzących obrót produktami leczniczymi.
- Dane te będą obowiązani przekazywać również przedstawiciele medyczni lub handlowi odwiedzający osoby uprawnione do wystawiania recept lub osoby prowadzące obrót produktami leczniczymi.
- Projekt w sposób ogólny opisuje tryb zmiany decyzji refundacyjnych dotyczących programów lekowych. W tym zakresie nie zostało doprecyzowane w jaki sposób będzie następowała ta zmiana.
- Brak jest przepisów przejściowych dotyczących odejścia od kontraktów zawieranych między aptekami a NFZ na rzecz relacji opartej wprost na przepisach ustawowych.
_________________________
1 Zob. http://blog.dzp.pl/pharma/pierwsza-nowelizacja-ustawy-o-refundacji/